Projects in our lab are connected by their shared motivation to understand gene regulation in human tissues and cells in health and disease. We employ a range of technologies and systems to achieve this goal.
Developing single-cell and single-molecule genomics approaches to probe gene regulation in unprecedented detail
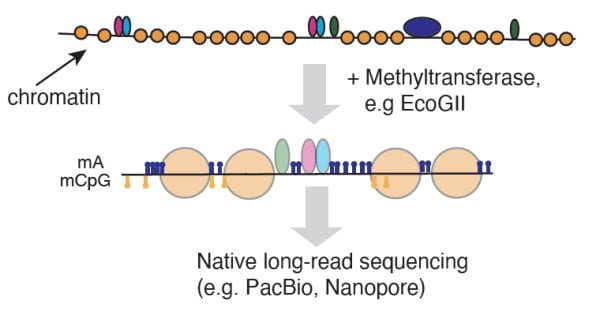
Schematic of mapping single-molecule chromatin organization using an m6A methyltransferase followed by long-read sequencing.
We are developing multimodal assays which simultaneously probe multiple molecular features in single cells and on individual molecules. These methods will allow us to identify and characterize cell types and gene regulatory states at a finer resolution than enabled by previously developed technologies. These methods will improve our understanding of how regulatory features work together in concert to coordinate cellular activity.We previously developed single-cell Nucleosome Occupancy and Methylation sequencing (scNOMe-seq), an assay which simultaneously measures chromatin accessibility and DNA methylation (Pott 2017). scNOMe-seq uses a GpC DNA methyltransferase to map chromatin accessibility. Our current projects build on this principle: we are combining methyltransferase-based methods with direct native sequence platforms such as PacBio with to map chromatin organization and protein-DNA interaction along continuous DNA fragments. We envision that these multimodal methods will become a toolbox to allow researchers to study regulatory processes in healthy and diseased tissues with unprecedented detail.
Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife, 6, e23203. (2017). https://doi.org/10.7554/eLife.23203.001.
This work is supported by an R35/MIRA from NIGMS and an R21 from NHGRI (with Kohta Ikegami), respectively.
Genetic and epigenetic disruption of cell-type-specific gene regulatory programs in IBD
Inflammatory bowel diseases (IBD), including Crohn’s Disease (CD) and Ulcerative Colitis (UC), result from a complex interaction between genetic, microbial, and environmental factors. However, specific mechanisms of disease initiation and progression remain mostly unknown. A major roadblock for progress has been the clinical heterogeneity of IBD, the highly variable nature of microbial populations, and the cellular complexity of the intestinal mucosa.
In collaboration with the groups of Oni Basu and Gene Change we are creating a cellular atlas of intestinal mucosa in healthy individuals and CD patients.
We are using this gut cell atlas to characterize cell-type-specific transcriptional and epigenetic states and to identify regulatory elements associated with inflammation in CD. In addition, cell-type-resolved regulatory elements help us to map putative causal non-coding variants from IBD GWAS and to identify their likely cell type(s) of action.
Building on these rich data sets, we will combine intestinal organoid cultures and high-throughput CRISPR-based perturbation experiments to mechanistically characterize the functional impact of regulatory elements and genetic variants associated with IBD in cell types of the epithelium and the lamina propria. Detailed mechanistic studies will be performed e.g., for epithelial risk variants/loci using organoids and single-molecule epigenetic assays.
Matthias Zilbauer, Kylie R James, Mandeep Kaur, Sebastian Pott, Zhixin Li, Albert Burger, et al. . A Roadmap for the Human Gut Cell Atlas. Nat Rev Gastroenterol Hepatol (2023). https://doi.org/10.1038/s41575-023-00784-1.
This work on this project is supported by a Gut Cell Atlas grant from the Helmsley Trust and partially through a R35/MIRA from NIGMS.
Identification and characterization of non-coding causal variants in atrial fibrillation (AF)
Genome-wide association studies (GWAS) have identified thousands of loci for hundreds of diseases and traits, promising to provide biological insights into complex traits by identifying putative disease genes and pathways. However, most GWAS signals are located in non-coding regions, making translating GWAS implication into molecular mechanisms underlying disease risk challenging.
The predominant paradigm is that genetic variants (SNPs) affect regulatory DNA, causing quantitative expression changes in target genes. However, GWAS data cannot identify causative SNPs, the gene target of the genetic variation, or in which cell-type gene regulation is affected. In an attempt to address these challenges, we performed single-cell epigenomic mapping (scATAC-seq) in human heart samples and used the data to enhance fine-mapping and to identify risk genes. This study provided a comprehensive map of AF risk variants and genes, and might be used more generally to integrate single-cell genomics with genetic studies of complex traits.
In our ongoing work we aim to extend this initial study to refine variant annotation and identification and to improve the the translation of AF GWAS signals into actionable biological mechanism using functional high-throughput assays.
Selewa A., Luo K., Wasney M., Smith L., Sun X., Tang C., Eckart H., Moskowitz IP, Basu A., He X., Pott S. (2023) Single-cell genomics improves the discovery of risk variants and genes of Atrial Fibrillation. Nat Commun. 2023 Aug 17;14(1):4999. https://doi.org/10.1101/2022.02.02.22270312
This work was initiated as part of a CZI funded Human Cell Atlas project. The current work is funded through an R01 from NHLBI with Ivan Moskowitz and Xin He.